
Review Article
Enhancing the Potency of F508del Correction: A Multi-Layer Combinational Approach to Drug Discovery for Cystic Fibrosis
With better understanding of the cellular and molecular pathophysiology underlying cystic fibrosis (CF), novel drugs are being developed that specifically target the molecular defects of the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-activated chloride channel on the plasma membrane that causes CF. Starting with cell-based high-throughput screening, small molecules have been identified that are able to fix specific molecular defects of various disease-causing CFTR mutants. With the successful development of ivacaftor, a “potentiator” that enhances CFTR chloride channel activity, new types of small-molecule compounds that “correct” the misfolding and misprocessing of the most common CF-causing mutation, F508del, are actively being sought for. Recent studies focused on the potential mechanisms of action of some of the investigational CFTR “correctors” shed new light on how the F508del mutant can be targeted in an attempt to ameliorate the clinical symptoms associated with CF. A multi-layer combinational approach has been proposed to achieve the highpotency correction necessary for significant clinical outcome. The mechanistic insights obtained from such studies will shape the future therapeutics development for the vast majority of CF patients.
Keywords
CFTR; Cystic fibrosis; Drug discovery; F508del; High-throughput screen; Molecular chaperones; Protein misfolding
INTRODUCTION
Cystic fibrosis and CFTR
Cystic fibrosis (CF) occurs due to mutation(s) in the cystic fibrosis transmembrane conductance regulator (CFTR) gene [1]. CFTR is a cAMP-regulated Cl- channel that functions at the apical surface of epithelia from the lung, intestines, pancreas, liver, and a number of other organs. The impaired salt and water transport leads to a multi-system disease, with the CF lung disease being the primary cause of mortality.
Current treatments of CF largely focus on ameliorating its various clinical symptoms. Antibiotics are administered to control lung infection. Hypertonic saline is used to reverse airway surface dehydration, pancreatic enzymes and vitamin supplements are used to correct pancreatic insufficiency, and liver/lung transplantation is performed to treat the end-stage organ failure. Therapeutics directed to specific molecular defects of CFTR mutants have the potential of simultaneously alleviating symptoms of all affected organs. Recently, the first such drug, ivacaftor, was approved by the U.S. Food and Drug Administration (FDA) to treat roughly 4% of CF patients that carry a G551D mutation in CFTR.
F508del defects
Over 90% of CF patients carry at least one allele of CFTR gene with the deletion of a phenylalanine at residue 508 (F508del). This mutation impairs the folding of newly synthesized CFTR in the endoplasmic reticulum (ER), and, as a result, dramatically reduces the transport of nascent CFTR from the ER to the Golgi [2], leading to loss of function at the cell surface. The mutant protein accumulates in the ER and is subjected to proteasome degradation [3, 4]. Aside from the trafficking defect, F508del mutant has impaired chloride channel activity characterized by reduced open probability [5]. Moreover, the small fraction of F508del CFTR that does reach the plasma membrane displays reduced stability at the cell surface, and is quickly cleared through the lysosomal pathway [6].
CFTR is a member of the ATP-binding cassette (ABC) family of solute transporters, and is made up of two halves, each containing a transmembrane domain (TMD) and a nucleotide- binding domain (NBD) [1] (Figure 1A). The two halves are connected by a regulatory (R) domain. The F508 residue resides in NBD1, and therefore NBD1 is the origin of F508del misfolding. F508del alters the folding kinetics of isolated NBD1 [7]. Recent data support a critical role for NBD1 in F508del misfolding during CFTR translation [8]. F508del mutation causes the solvent exposure of a buried, hydrophobic residue (V510), leading to altered surface topography of NBD1 and its aberrant interactions with other domains [9-11] (Figure 1B). The end result is multi- domain misfolding, and the rescue of such global misfolding is necessary for effective F508del correction [12,13].
Figure 1 F508del: misfolding, rescue and drug discovery strategy. (A) wild-type CFTR conformation. (B) F508del CFTR conformation. (C) Small-molecule modulators of F508del CFTR and their impact on its conformation. I, Class I corrector; II, Class II corrector; CR, chaperone regulator; NS, NBD1 stabilizer; P, potentiator; and SS, surface stabilizer. (D) Multi-layer combinational strategy for the development of high-potency F508del correctors.
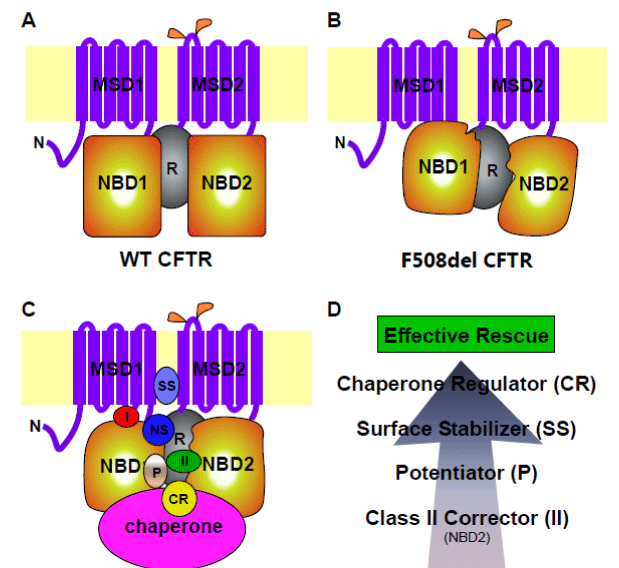
Figure 1 F508del: misfolding, rescue and drug discovery strategy. (A) wild-type CFTR conformation. (B) F508del CFTR conformation. (C) Small-molecule modulators of F508del CFTR and their impact on its conformation. I, Class I corrector; II, Class II corrector; CR, chaperone regulator; NS, NBD1 stabilizer; P, potentiator; and SS, surface stabilizer. (D) Multi-layer combinational strategy for the development of high-potency F508del correctors.
×
High-througput screening and the initial success
The systematic search for small molecule compounds that are able to modify the folding and/or activity of mutant CFTR was first attempted by high-throughput screenings (HTS), where relatively simple cell-based assays were used to select compounds that are capable of exerting a significant effect on the functional expression of specific CFTR mutants at the cell surface. Early assays used for such screenings are largely based on CFTR-mediated chloride channel activity.
Iodide efflux assay
Early in 1999, iodide efflux assay was used to evaluate the ability of certain small molecule compounds to activate the chloride channel activity of CFTR, leading to the identification of benzo(c)quinolizinium compounds (e.g. MPB-07, MPB-27, and MPB-91) as CFTR activators [14,15]. Surprisingly, MPB-07 and MPB-91 were also found to facilitate F508del processing [16]. The concentrations of the compounds used were ~ 250µM. At the same concentration, both compounds were found to inhibit the degradation of F508del-harboring NBD1-R domains [17]. These data provided the earliest evidence that certain CFTR potentiators can have corrector activities.
Iodide-sensitive YFP assay
The Verkman's group at the University of California San Francisco utilized an iodide influx assay employing halide sensitive yellow fluorescent protein (YFP), where the rate of YFP quenching by extracellular iodide is measured to determine the CFTR activity [18]. Using this assay, compounds that promote the cell surface functional expression of F508del CFTR can be identified. Such compounds can either promote the processing of F508del CFTR (correctors), or specifically enhance its chloride channel activity (potentiators), or perhaps both. To specifically search for CFTR potentiators, cells stably expressing F508del CFTR were subjected to low temperature rescue to mobilize the mutant protein to the cell surface prior to compound treatment. Using the above approaches, both F508del correctors (e.g. corr-4a and corr-2b) and potentiators have been identified [18-20].
Voltage-sensitive FRET assay
Vertex Pharmaceuticals used a voltage-sensitive, fluorescence resonance energy transfer (FRET) system to measure anion flux through CFTR [21]. This latter approach led to the development of a potentiator (VX-770) that enhances the channel gating of CFTR carrying a G551D mutation [22]. In 2012, VX-770 was approved by the U.S. FDA as an oral drug under the name of Kalydeco (ivacaftor) for treatment of CF patients six years or older, who carry the G551D mutant. The successful development of the first CFTR modulator for the treatment of ~4% of CF patients provides the first proof-of-principle for the therapeutic rationale that CF patients can be effectively treated by correcting the specific molecular defect of CFTR using a small molecule.
Using the same assay, Vertex also identified a series of F508del correctors including VRT-325 [21] and VX-809 [23]. The latter, in combination with ivacaftor, is currently under clinical trial for the treatment of CF patients carrying the F508del mutation.
CFTR trafficking assay
Complementing the assays based on CFTR chloride channel activity, the Thomas group at McGill University developed an assay that is based on the cell surface localization of the F508del CFTR by inserting three copies of hemagglutinin into the fourth extracellular loop of CFTR. The F508del CFTR on the cell surface was detected by immunofluorescence staining [24]. Using this assay, a number of correctors were identified including sildenafil, a phosphodiesterase inhibitor [24].
NBD1 melting assay
As F508del impairs the folding of CFTR NBD1, another assay was developed to assess the thermal stability of F508del NBD1 using differential scanning fluorimetry. Starting from 224 primary hits from a HTS using the CFTR trafficking assay, the NBD1 melting assay was used to select compounds that will improve the thermal stability of F508del NBD1, and only one compound was identified, the RDR1 [25].
Domain-interface-based virtual screen
Given that the F508del mutation causes CFTR misfolding through interfering with domain- domain interactions, targeting critical domain interfaces might aid in the identification of new correctors. Scientists at EPIX Pharmaceuticals developed three in silico screening regimes focusing on the NBD1-NBD2 interface, the NBD1-ICL4 interface, and the NBD1:NBD2:ICL1:ICL2:ICL4 multiple domain interface [26]. The 496 preliminary hits were then subjected to a second round of HTS using the iodide flux assay [18], yielding a total of 15 correctors [26].
NBD1-based virtual screen
Using Molecular Dynamics simulation, a small molecule binding cleft was identified in F508del NBD1. The occupation of the cleft by small molecules inhibits the unfolding of the domain. Using virtual screen and fragment based molecular design, a series of small molecule compounds were generated that modestly enhanced F508del CFTR processing in the cell [27].
Targeted compound libraries
Using the iodide-sensitive YFP assay, a library of kinase inhibitors that are currently in clinical use or in clinical trials for treating other diseases including cancer and inflammatory diseases were screened for functional correction of F508del CFTR [28]. Several of these kinase inhibitors were identified to enhance the functional expression of F508del CFTR at the cell surface. While the mechanisms of such correction remain evasive at the present time, the utilization of compound libraries that are already in clinical use or are under clinical trials will potentially accelerate the advancement of promising compounds along the CF drug discovery pipeline.
Mechanisms of correction
During the past few years, significant progress has been made in understanding the mechanisms of action of some of the existing investigational correctors. Such understanding provides an initial conceptual framework through which rational efforts can be made to enhance the effectiveness of future CF drug development.
VRT-325
The first intensively studied F508del corrector is VRT-325 (also known as Cor-325) developed by Vertex Pharmaceuticals. VRT-325 was found to rescue not only F508del CFTR but also other CFTR processing mutants such as R258G, S945L, and H949Y, and processing mutants of P-glycoprotein (P-gp), a drug pump that also belongs to the ABC transporter family [29]. Interestingly, VRT-325 also inhibits P-gp transport activity [29], suggesting that Cor-325 targets certain common structural features of ABC transporters. In contrast, corr-2b, a corrector developed by the Verkman group at University of California San Francisco, specifically rescues CFTR processing mutants but not P-gp processing mutants [30], suggesting that corr-2b targets a structural feature that is unique to CFTR and not shared by other ABC transporters.
In a separate study, in situ limited proteolysis was employed to assess the impact of VRT-325 on domain conformation in the context of membrane-bound, full-length CFTR [31]. VRT-325 enhances the protease resistance of NBD1 but not that of other domains in F508del CFTR. In contrast, combined second-site suppressor mutations R553K/R555K (2RK) in NBD1 not only alter the protease susceptibility of NBD1 but also that of other domains of F508del CFTR. These data suggest that VRT-325 exerts its effect through NBD1. While both VRT-325 and the 2RK mutations only lead to partial rescue of F508del misprocessing separately, the combination of the two brings the F508del processing to close to wild-type level. Despite the virtually full restoration of F508del processing, its NBD1 conformational defect was not fully corrected according to its protease susceptibility. In parallel to this, a defect in channel gating persists, which was fully corrected by the potentiator ivacaftor. This study points to the possibility that distinct conformational features in NBD1 and perhaps even in full-length CFTR contribute to F508del misprocessing and its defective channel activity, and therefore combination of a corrector and a potentiator has the potential of providing maximal functional correction of F508del defects.
VX-809
VX-809, an investigational corrector developed by Vertex Pharmaceuticals, restores F508del CFTR functional expression to ~14% of wild-type CFTR in human bronchial epithelial cells [23]. VX-809 was able to rescue F508del CFTR in the absence of NBD2 and R domain, suggesting that these domains are dispensable for VX-809-mediated rescue [32]. In the same study, VX-809 was found to specifically stabilize purified, recombinant TMD1 but not NBD1 or TMD2. These results suggest that VX-809 rescues F508del through CFTR TMD1.
Mechanism-based corrector classification
A recent study conducted by the Lukacs group at McGill analyzed the impact of many correctors on different domains of F508del CFTR [33]. They were able to group various correctors based on their target domain(s) within CFTR. Class I correctors such as VX-809, VRT-325 (C3), and VRT-534 (C18) were found to stabilize the interfaces between NBD1 and MSD1/MSD2 (Figure 1C, “I”). Class II correctors such as Corr-4a (C4) and core-corr-II were found to target NBD2 (Figure 1C, “II”). Chemical chaperones were able to stabilize F508del NBD1. The combinational use of correctors from different classes dramatically enhances rescue potency. This study left a very important question: Is it possible to get a CFTR-specific NBD1 stabilizer? 100H620Q substitution at the C-terminal region of NBD1 is known to increase the channel open probability of CFTR [34]. CFFT-001 is a dual-acting corrector/potentiator developed by EPIX Pharmaceuticals. NMR studies of human F508del NBD1 either carry the H620Q mutation or treated with CFFT-001 revealed a similar disruption of the interactions between the β-strands S3, S9, and S10 and the C-terminal helices H8 and H9, suggesting a similar conformational shift [35]. Differential scanning calorimetry showed a reduced Tm of NBD1 in the presence of H620Q or CFFT-001, suggesting that CFFT-001 binds to a less stable conformation of NBD1 [35]. Taken together, these data suggest that small-molecule compounds that modulate F508del NBD1 conformation have the potential to simultaneously regulate CFTR folding and channel activity.
Rational approaches
Benzbromarone: an F508del stabilizer at the cell surface.
Aside from reduced maturation and subnormal channel activity, F508del has reduced stability at the cell surface. Studies on P-gp have revealed that the presence of substrate molecules during the synthesis of P-gp folding/trafficking mutants enhances their maturation [36]. Therefore, small molecule substrates can serve as chemical chaperones to facilitate the maturation and stabilization of misfolded P-gp mutants. Further studies indicated that the TMDs of P-gp interact with the substrates, and are hence responsible for the enhanced folding and maturation of the P-gp mutants [37]. Several P-gp inhibitors including benzbromarone also inhibit CFTR chloride channel activity by blocking the pore [38]. Interestingly, benzbromarone was recently found to enhance F508del CFTR processing by stabilizing it at the cell surface [39] (Figure 1C, “SS”, surface stabilizer). Future studies in this direction might reveal novel correctors that function through stabilizing F508del native conformation, and hence improve maturation, cell surface stability, and even channel activity of F508del CFTR.
Modulators of chaperone machinery
The folding, maturation and cell surface stabilization of CFTR within the cell occur in the context of a huge network of molecular chaperones organized on both sides of the ER membrane [40]. The level and functionality of these chaperone proteins have a significant impact on F508del rescue. Several small-molecule compounds have the ability to regulate the expression and/or functionality of molecular chaperones, and therefore are able to promote the folding of F508del CFTR (Fig. 1C, “CR”, chaperone regulator).
Global chaperone regulators: Many molecular chaperones are regulated by the heat-shock response. Cetastrol, a small molecule inducer of the heat-shock response [41], was found to stabilize F508del CFTR and promote its rescue at reduced temperature [42]. Transcription regulator 4-phenylbutyrate is an oral drug recently approved for treating urea cycle disorders. It promotes F508del CFTR maturation and function in CF airway epithelial cells [43]. This compound was found to regulate the expression of a large number of proteins involved in protein folding, trafficking and degradation [44, 45]. Histone deacetylase inhibitor suberoylanilide hydroxamic acid, which impacts the expression of a large array of genes involved in numerous cellular processes, was found to increase the cell surface functional expression of F508del CFTR in CF airway epithelial cells [46]. Such “global” chaperone regulators have the capacity of enhancing F508del CFTR folding but their potential off-target effects might significantly limit their usefulness.
Doxorubicin: A cancer drug was found to increase the cell surface expression and chloride secretion of F508del CFTR [47]. Doxorubicin acts as a chemotherapy agent primarily by intercalating DNA and hence inhibiting its synthesis [48]. A recent study suggested that doxorubicin reduces the association of F508del CFTR with molecular chaperone Hsp70, and stabilizes it from accelerated degradation, suggesting that doxorubicin might rescue F508del CFTR through altering its interaction with cytoplasmic chaperones [49].
S-nitrosoglutathione (GSNO): Is an endogenous bronchodilator, whose concentration is low in CF airways [50]. GSNO was found to promote F508del processing [51] through a number of mechanisms involving specific chaperones such as cysteine strong proteins [52] and Hsp70/Hsp90 organizing protein [53].
Miglustat: The N-linked glycosylation of CFTR allows the interaction of nascent CFTR with the ER luminal chaperone calnexin, which retains misfolded or incompletely assembled CFTR from exiting the ER [54]. Miglustat, an α-1,2-glucosidase inhibitor that inhibits the deglucosylation of glycoproteins in the ER lumen, is used in the treatment of Gaucher disease. Miglustat was found to disrupt F508del CFTR interaction with calnexin in the ER lumen and partially restore the cAMP-activated chloride current of CF epithelial cells [55]. Miglustat improves both sodium and chloride transport defects of F508del CFTR in a human CF nasal epithelial cell line and in excised nasal epithelium of F508del/F508del transgenic mice [56,57]. Aside from its ability to correct the molecular defect of F508del CFTR, miglustat also reduces inflammatory response to P. aeruginosa, a serious bacterial threat especially to CF patients [58]. As demonstrated with miglustat, existing drugs for other diseases may benefit CF patients.
Current problems
The past decade witnessed tremendous advancement in our understanding of F508del misfolding and in the development of therapeutics that are directed at the core molecular defects of various CFTR mutants. One such drug has been approved by the FDA, and many investigational small molecule compounds are currently under clinical trials at various stages. The CF community is still anticipating the first FDA-approved drug that treats the F508del defect present in the vast majority of CF patients. Two of our major challenges are potency and toxicity.
In order to achieve clinically measurable improvements in CF patients, current investigational correctors need to rise above a potency threshold in preclinical development. As the F508del molecular defects are multifaceted, recent studies suggest that simultaneously fixing different molecular defects such as NBD1 misfolding and interdomain miscontact might be necessary for efficient correction [59,60]. This notion was echoed by a recent study suggesting that combinations of different classes of correctors can dramatically improve potency [33].
CFTR is a member of the ABC family of transporters. Some small molecules that bind to CFTR and alter its conformation also impact the conformation and function of other ABC transporters or even other ion channels [29]. This in turn might lead to high toxicity and other adverse effects. Correctors that are more specific to F508del CFTR are highly desirable. Many small-molecule regulators of chaperone machinery are known to improve F508del maturation [43,46,49,50,55]. However, chaperone regulators can have indirect effects on other physiologically important proteins, and therefore may lead to undesirable adverse effects. Such effects need to be carefully assessed prior to further development.
Future approaches
Structure-activity relationship
One potential way of improving potency for existing correctors is to perform structure-activity relationship studies in an attempt to optimize existing scaffolds. Certain cyanoquinolines were found to have independent corrector and potentiator activities [61]. Structure-activity relationship of these compounds revealed clear conformational properties of these dual action molecules necessary to elicit corrector and potentiator activities, respectively [62]. Among the small- molecule correctors identified from the Verkman screen [18], 4'-methyl-4, 5'-bithiazole appears to be highly promising for further development. 148 analogs were synthesized and subjected to structure-activity relationship studies, which revealed that the bithiazole substructure plays a critical function [63]. In fact, C17, a highly potent corrector, arose from such studies. As additional novel corrector scaffolds are identified through HTS, such medicinal chemistry “fine- tuning” is necessary to generate potent and safe correctors for clinical studies.
F508del NBD1 stabilizers
F508 resides in NBD1, and the impact of the F508del mutation on NBD1 folding and its subsequent impact on the folding of other CFTR domains are apparent [7,10,13,64-66]. Although the correction of the defects in both NBD1 conformation and domain-domain interactions are necessary for effective rescue [59,60], very few effective small-molecule correctors have been identified that specifically stabilize the mutant NBD1 [33] (Figure 1C, “NS”, NBD1 stabilizer). Novel HTS assays that focus on F508del NBD1 folding might fill this gap. It is worth noting that small-molecule potentiators often bind to NBDs [67] (Fig. 1C, “P”, potentiator). Therefore, it is possible that additional potentiators might arise from such a screen. Given the fact that F508del misfolding originates in NBD1, it is possible that a certain small-molecule compound might be able to bind to a critical site in the mutant NBD1 and alter its conformation in a manner that dramatically improves NBD1 conformation and its interactions with other domains, leading to efficient functional rescue of F508del CFTR at the cell surface.
A multi-layer combinational approach to effective F508del correction
Recent studies have indicated that combining correctors targeting different conformational defects of F508del leads to a much more effective rescue [33]. The identification of novel NBD1 stabilizers might further enhance the rescue potency of existing Class I and Class II correctors (Figure 1D). Potentiators and correctors appear to target distinct conformational elements in F508del CFTR [62]. The combination of the two will significantly enhance the cell surface functional expression of F508del CFTR (Figure 1D). In fact, the ivacaftor-VX-809 combination is currently under clinical trial for the treatment of F508del CF patients. Novel dual-function correctors-potentiators have been developed that can simultaneously enhance processing and channel activity of F508del CFTR in one small molecule [61]. The above combination can be further enhanced by adding a cell surface stabilizer for F508del such as Benzbromarone [39], and therefore ameliorating all three facets of the F508del defect and leading to high efficiency correction (Figure 1D). Once proven safe, an additional layer of enhancement in correction can be achieved through adding small-molecule chaperone regulators that improve the biological folding of F508del in the cell by increasing the foldable pool [68] and/or the foldability of the mutant protein (Fig. 1D). This multilayer combination of small-molecule modulators of F508del might eventually elevate the potency to a level that provides significant clinical outcome in the majority of CF patients (Figure 1D).
Acknowledgements
This work was supported by NIDDK/NIH grant P30DK072482, the Cystic Fibrosis Foundation Grant WANG12P0, and Samford University McWhorter Faculty Research Grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989; 245: 1066-1073.
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990; 63: 827-834.
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995; 83: 129-135.
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995; 83: 121-127.
- Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, et al. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature. 1991; 354: 526-528.
- Lukacs GL, Chang XB, Bear C, Kartner N, Mohamed A, Riordan JR, et al. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1993; 268: 21592-8.
- Qu BH, Strickland EH, Thomas PJ. Localization and suppression of a kinetic defect in cystic fibrosis transmembrane conductance regulator folding. J Biol Chem. 1997; 272: 15739-15744.
- Hoelen H, Kleizen B, Schmidt A, Richardson J, Charitou P, Thomas PJ, et al. The primary folding defect and rescue of ΔF508 CFTR emerge during translation of the mutant domain. PLoS One. 2010; 5: e15458.
- Du K, Sharma M, Lukacs GL. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol. 2005; 12: 17-25.
- Lewis HA, Wang C, Zhao X, Hamuro Y, Conners K, Kearins MC, et al. Structure and dynamics of NBD1 from CFTR characterized using crystallography and hydrogen/deuterium exchange mass spectrometry. J Mol Biol. 2010; 396: 406-430.
- Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, et al. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci U S A. 2008; 105: 3256-3261.
- Du K, Lukacs GL. Cooperative assembly and misfolding of CFTR domains in vivo. Mol Biol Cell. 2009; 20: 1903-1915.
- Roy G, Chalfin EM, Saxena A, Wang X. Interplay between ER exit code and domain conformation in CFTR misprocessing and rescue. Mol Biol Cell. 2010; 21: 597-609.
- Becq F, Mettey Y, Gray MA, Galietta LJ, Dormer RL, Merten M, et al. Development of substituted Benzo[c]quinolizinium compounds as novel activators of the cystic fibrosis chloride channel. J Biol Chem. 1999; 274: 27415-27425.
- Dérand R, Bulteau-Pignoux L, Mettey Y, Zegarra-Moran O, Howell LD, Randak C, et al. Activation of G551D CFTR channel with MPB-91: regulation by ATPase activity and phosphorylation. Am J Physiol Cell Physiol. 2001; 281: C1657-1666.
- Dormer RL, Dérand R, McNeilly CM, Mettey Y, Bulteau-Pignoux L, Métayé T, et al. Correction of delF508-CFTR activity with benzo(c)quinolizinium compounds through facilitation of its processing in cystic fibrosis airway cells. J Cell Sci. 2001; 114: 4073-4081.
- Stratford FL, Pereira MM, Becq F, McPherson MA, Dormer RL. Benzo(c)quinolizinium drugs inhibit degradation of Delta F508-CFTR cytoplasmic domain. Biochem Biophys Res Commun. 2003; 300: 524-530.
- Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, et al. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005; 115: 2564-2571.
- Pedemonte N, Sonawane ND, Taddei A, Hu J, Zegarra-Moran O, Suen YF, et al. Phenylglycine and sulfonamide correctors of defective delta F508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol Pharmacol. 2005; 67: 1797-807.
- Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, et al. Nanomolar affinity small molecule correctors of defective Delta F508-CFTR chloride channel gating. J Biol Chem. 2003; 278: 35079-35085.
- Van Goor F, Straley KS, Cao D, González J, Hadida S, Hazlewood A, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006; 290: L1117-1130.
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009; 106: 18825-18830.
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011; 108: 18843-18848.
- Carlile GW, Robert R, Zhang D, Teske KA, Luo Y, Hanrahan JW, et al. Correctors of protein trafficking defects identified by a novel high-throughput screening assay. Chembiochem. 2007; 8: 1012-1020.
- Sampson HM, Lam H, Chen PC, Zhang D, Mottillo C, Mirza M, et al. Compounds that correct F508del-CFTR trafficking can also correct other protein trafficking diseases: an in vitro study using cell lines. Orphanet J Rare Dis. 2013; 8: 11.
- Kalid O, Mense M, Fischman S, Shitrit A, Bihler H, Ben-Zeev E, et al. Small molecule correctors of F508del-CFTR discovered by structure-based virtual screening. J Comput Aided Mol Des. 2010; 24: 971-991.
- Pasyk EA, Foskett JK. Mutant (delta F508) cystic fibrosis transmembrane conductance regulator Cl- channel is functional when retained in endoplasmic reticulum of mammalian cells. J Biol Chem. 1995; 270: 12347-12350.
- Trzcinska-Daneluti AM, Nguyen L, Jiang C, Fladd C, Uehling D, Prakesch M, et al. Use of kinase inhibitors to correct ΔF508-CFTR function. Mol Cell Proteomics. 2012; 11: 745-757.
- Loo TW, Bartlett MC, Clarke DM. Rescue of DeltaF508 and other misprocessed CFTR mutants by a novel quinazoline compound. Mol Pharm. 2005; 2: 407-413.
- Wang Y, Bartlett MC, Loo TW, Clarke DM. Specific rescue of cystic fibrosis transmembrane conductance regulator processing mutants using pharmacological chaperones. Mol Pharmacol. 2006; 70: 297-302.
- Yu W, Kim Chiaw P, Bear CE. Probing conformational rescue induced by a chemical corrector of F508del-cystic fibrosis transmembrane conductance regulator (CFTR) mutant. J Biol Chem. 2011; 286: 24714-24725.
- Loo TW, Bartlett MC, Clarke DM. Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem Pharmacol. 2013; 86: 612-619.
- Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, et al. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat Chem Biol. 2013; 9: 444-454.
- Billet A, Melin P, Jollivet M, Mornon JP, Callebaut I, Becq F. C terminus of nucleotide binding domain 1 contains critical features for cystic fibrosis transmembrane conductance regulator trafficking and activation. J Biol Chem. 2010; 285: 22132-40.
- Hudson RP, Chong PA, Protasevich II, Vernon R, Noy E, Bihler H, et al. Conformational changes relevant to channel activity and folding within the first nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2012; 287: 28480-94.
- Loo TW, Clarke DM. Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. J Biol Chem. 1997; 272: 709-712.
- Loo TW, Clarke DM. The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J Biol Chem. 1999; 274: 24759-24765.
- Diena T, Melani R, Caci E, Pedemonte N, Sondo E, Zegarra-Moran O, et al. Block of CFTR-dependent chloride currents by inhibitors of multidrug resistance-associated proteins. Eur J Pharmacol. 2007; 560: 127-131.
- Loo TW, Bartlett MC, Clarke DM. Benzbromarone stabilizes ΔF508 CFTR at the cell surface. Biochemistry. 2011; 50: 4393-4395.
- Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006; 127: 803-815.
- Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S, et al. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem. 2004; 279: 56053-56060.
- Wang X, Koulov AV, Kellner WA, Riordan JR, Balch WE. Chemical and biological folding contribute to temperature-sensitive DeltaF508 CFTR trafficking. Traffic. 2008; 9: 1878-1893.
- Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest. 1997; 100: 2457-2465.
- Singh OV, Vij N, Mogayzel PJ Jr, Jozwik C, Pollard HB, Zeitlin PL. Pharmacoproteomics of 4-phenylbutyrate-treated IB3-1 cystic fibrosis bronchial epithelial cells. J Proteome Res. 2006; 5: 562-71.
- Choo-Kang LR, Zeitlin PL. Induction of HSP70 promotes DeltaF508 CFTR trafficking. Am J Physiol Lung Cell Mol Physiol. 2001; 281: L58-68.
- Hutt DM, Herman D, Rodrigues AP, Noel S, Pilewski JM, Matteson J, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol. 2010; 6: 25-33.
- Maitra R, Shaw CM, Stanton BA, Hamilton JW. Increased functional cell surface expression of CFTR and DeltaF508-CFTR by the anthracycline doxorubicin. Am J Physiol Cell Physiol. 2001; 280: C1031-1037.
- Fornari FA, Randolph JK, Yalowich JC, Ritke MK, Gewirtz DA. Interference by doxorubicin with DNA unwinding in MCF-7 breast tumor cells. Mol Pharmacol. 1994; 45: 649-656.
- Maitra R, Hamilton JW. Altered biogenesis of deltaF508-CFTR following treatment with doxorubicin. Cell Physiol Biochem. 2007; 20: 465-472.
- Grasemann H, Gaston B, Fang K, Paul K, Ratjen F. Decreased levels of nitrosothiols in the lower airways of patients with cystic fibrosis and normal pulmonary function. J Pediatr. 1999; 135: 770-772.
- Andersson C, Gaston B, Roomans GM. S-Nitrosoglutathione induces functional DeltaF508-CFTR in airway epithelial cells. Biochem Biophys Res Commun. 2002; 297: 552-557.
- Zaman K, Carraro S, Doherty J, Henderson EM, Lendermon E, Liu L, et al. S-nitrosylating agents: a novel class of compounds that increase cystic fibrosis transmembrane conductance regulator expression and maturation in epithelial cells. Mol Pharmacol. 2006; 70: 1435-42.
- Marozkina NV, Yemen S, Borowitz M, Liu L, Plapp M, Sun F, et al. Hsp 70/Hsp 90 organizing protein as a nitrosylation target in cystic fibrosis therapy. Proc Natl Acad Sci U S A. 2010; 107: 11393-11398.
- Pind S, Riordan JR, Williams DB. Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1994; 269: 12784-12788.
- Norez C, Noel S, Wilke M, Bijvelds M, Jorna H, Melin P, et al. Rescue of functional delF508-CFTR channels in cystic fibrosis epithelial cells by the alpha-glucosidase inhibitor miglustat. FEBS Lett. 2006; 580: 2081-2086.
- Noël S, Wilke M, Bot AG, De Jonge HR, Becq F. Parallel improvement of sodium and chloride transport defects by miglustat (n-butyldeoxynojyrimicin) in cystic fibrosis epithelial cells. J Pharmacol Exp Ther. 2008; 325: 1016-1023.
- Norez C, Antigny F, Noel S, Vandebrouck C, Becq F. A cystic fibrosis respiratory epithelial cell chronically treated by miglustat acquires a non-cystic fibrosis-like phenotype. Am J Respir Cell Mol Biol. 2009; 41: 217-225.
- Dechecchi MC, Nicolis E, Norez C, Bezzerri V, Borgatti M, Mancini I, et al. Anti-inflammatory effect of miglustat in bronchial epithelial cells. J Cyst Fibros. 2008; 7: 555-565.
- Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, et al. Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell. 2012; 148: 150-163.
- Mendoza JL, Schmidt A, Li Q, Nuvaga E, Barrett T, Bridges RJ, et al. Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell. 2012; 148: 164-174.
- Phuan PW, Yang B, Knapp JM, Wood AB, Lukacs GL, Kurth MJ, et al. Cyanoquinolines with independent corrector and potentiator activities restore ΔPhe508-cystic fibrosis transmembrane conductance regulator chloride channel function in cystic fibrosis. Mol Pharmacol. 2011; 80: 683-93.
- Knapp JM, Wood AB, Phuan PW, Lodewyk MW, Tantillo DJ, Verkman AS, et al. Structure-activity relationships of cyanoquinolines with corrector-potentiator activity in ΔF508 cystic fibrosis transmembrane conductance regulator protein. J Med Chem. 2012; 55: 1242-51.
- Yoo CL, Yu GJ, Yang B, Robins LI, Verkman AS, Kurth MJ. 4'-Methyl-4,5'-bithiazole-based correctors of defective delta F508-CFTR cellular processing. Bioorg Med Chem Lett. 2008; 18: 2610-2614.
- He L, Aleksandrov LA, Cui L, Jensen TJ, Nesbitt KL, Riordan JR. Restoration of domain folding and interdomain assembly by second-site suppressors of the DeltaF508 mutation in CFTR. FASEB J. 2010; 24: 3103-3112.
- Thibodeau PH, Richardson JM 3rd, Wang W, Millen L, Watson J, Mendoza JL, et al. The cystic fibrosis-causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J Biol Chem. 2010; 285: 35825-35835.
- Thomas PJ, Ko YH, Pedersen PL. Altered protein folding may be the molecular basis of most cases of cystic fibrosis. FEBS Lett. 1992; 312: 7-9.
- Zegarra-Moran O, Monteverde M, Galietta LJ, Moran O. Functional analysis of mutations in the putative binding site for cystic fibrosis transmembrane conductance regulator potentiators. Interaction between activation and inhibition. J Biol Chem. 2007; 282: 9098-104.
- Younger JM, Ren HY, Chen L, Fan CY, Fields A, Patterson C, et al. A foldable CFTR{Delta}F508 biogenic intermediate accumulates upon inhibition of the Hsc70-CHIP E3 ubiquitin ligase. J Cell Biol. 2004; 167: 1075-1085.
Cite this article: Kirby EF, Heard AS, Wang XR (2013) Enhancing the Potency of F508del Correction: A Multi-Layer Combinational Approach to Drug Discovery for Cystic Fibrosis. J Pharmacol Clin Toxicol 1(1): 1007.
Current Issue Vol.1.1
The Redox Brain and Nitric Oxide: Implications for Psychiatric Illness
Caroline E Wass1 and Ana Andreazza2,3*
Caroline E Wass1 and Ana Andreazza2,3*
Pharmacological Modulators of Endothelial Progenitor Cell Therapy: Implications for Treatment with Thiazolidinediones
Maria Korah1, Yagna PR Jarajapu2, Maria B. Grant3 and Ashay D. Bhatwadekar3*
Maria Korah1, Yagna PR Jarajapu2, Maria B. Grant3 and Ashay D. Bhatwadekar3*
Profile of Suspect Adverse Drug Reactions in a Teaching Tertiary Care Hospital
Shubhatara Swamy1, Bhanuprakash2, Pratibha Nadig1*, Muralimohan3 and Manjula Shetty1
Shubhatara Swamy1, Bhanuprakash2, Pratibha Nadig1*, Muralimohan3 and Manjula Shetty1
Enhancing the Potency of F508del Correction: A Multi-Layer Combinational Approach to Drug Discovery for Cystic Fibrosis
Emily F Kirby, Ashley S Heard and X Robert Wang*
Emily F Kirby, Ashley S Heard and X Robert Wang*
Leptin Up-Regulates HECTD1 to Promote Phosphoinositide Metabolism and Cell Migration and Invasion in Breast Cancer Cells
Qiaodan Zheng1,2#, Xiang Li2#, Manjula Sunkara3, Andrew J. Morris3, Wenyuan Wu1* and Cai Huang2,4*
Qiaodan Zheng1,2#, Xiang Li2#, Manjula Sunkara3, Andrew J. Morris3, Wenyuan Wu1* and Cai Huang2,4*
Copyright © 2013 JSciMed Central. All rights reserved.