
Review Article
Integration of AKT and ERK Signaling Pathways in Cancer: Biological and Therapeutic Implications
The PI3K/AKT and RAS/RAF/MEK/ERK signaling pathways are often activated concurrently by separate genetic alterations in human cancer. Although the selective advantage of mutational activation of both pathways remains largely elusive, emerging evidences indicate that both pathways interact to regulate each other and cooperate to maintain the transformed phenotype and promote cancer progression and metastasis. Here, we focus on recent findings on the convergent regulation of downstream functions by AKT and ERK pathways and discuss the biologic and therapeutic relevance of the pathway convergence in cancer.
Keywords
AKT; ERK; Cross-talk; Pathway convergence; 4E-BP1; BAD; Cap-dependent translation; Cancer progression; Metastasis
Introduction
Oncogenic activation of AKT and ERK signaling pathways
The PI3K/AKT and RAS/RAF/MEK/ERK pathways are the central signal transduction mechanisms for controlling cell proliferation, survival, metabolism and motility in response to extracellular stimuli. Mutations in genes that encode components of these two pathways occur at high frequency in cancer. The former pathway is often activated in a majority of human cancers due to the activating mutations of the catalytic subunit of PI3K p110α(PIK3CA) and the inactivating mutations or decreased function of PTEN, whereas hyperactivation of MEK/ERK signaling driven by mutant RAS and BRAF is also a common oncogenic event in a variety of cancers [1,2]. Moreover, the concurrent activation of both the AKT and ERK pathways by separate mutations occurs in a significant portion of human tumors. For instance, KRAS and PIK3CA mutation; BRAF and PIK3CA mutation; and BRAF and PTEN mutation occur simultaneously in colorectal carcinoma, thyroid carcinoma and melanoma, respectively [3-7]. Growing evidences indicate that the mutationally-activated AKT and ERK signaling pathways cooperate to promote cancer progression and metastasis, which is associated with poor outcomes and tumor recurrence.
Cross-talk between AKT and ERK pathways
The prevalence of the AKT and ERK pathway activation in human cancers has led to the aggressive development of PI3K, AKT, RAF and MEK inhibitors as anticancer drugs. Preclinical studies and clinical trials with selective PI3K and AKT inhibitors show that mutant PIK3CA tumors tend to be dependent on the PI3K/AKT pathway and sensitive to the pathway inhibition [8-10]. On the other hand, the BRAF inhibitor vemurafenib or the MEK inhibitor trametinib produces high response rates in melanoma patients whose tumors harbor the BRAF-V600E activating mutation [11,12]. However, resistance to these targeted drugs generally is observed within a few months, and tumors eventually regrow and progress in almost all patients [7,13].
Resistance to targeted inhibitors is an emerging problem with varied mechanisms. We and others have found that tumor cells with PIK3CA or PTEN mutation are not all sensitive to the inhibitors of PI3K or AKT [14,15]. Similarly, mutant KRAS or BRAF tumors are not always dependent on the ERK signaling and sensitive to the BRAF and MEK inhibitors [15-17]. We demonstrate that coexistent KRAS mutation renders PIK3CA mutant tumors independent of PI3K/AKT signaling, whereas PIK3CA mutation uncouples tumor growth from MEK/ERK and mutant KRAS signaling [15,17]. In tumors with concurrent mutational activation of both PI3K/AKT and MEK/ERK pathways, inhibiting either pathway alone has minor or negligible effects on tumor growth and cell survival and motility. Combined inhibition of both pathways is required to effectively induce apoptosis and inhibit cell motility and tumor growth [15,18]. We have attributed this requirement to the existence of downstream proteins that integrate the oncogenic functions of AKT and ERK signaling pathways in tumor progression and metastasis [15,18] (Figure 1). Coexisting mutational activation of the two pathways independently contributes to tumor growth and metastasis by convergent regulation of function of these integrators, thus reducing 'oncogenic addiction' on AKT or ERK signaling pathway and causing the tumor resistance to inhibition of either pathway.
Figure 1 A cross-talk between the PI3K/AKT and RAS/RAF/MEK/ERK signaling pathways via convergent regulation of their common downstream targets for cancer progression. We have identified 4E-BP1 and BAD as key effectors of the oncogenic activation of both pathways that integrate their function in tumorigenesis and metastasis. *mutation
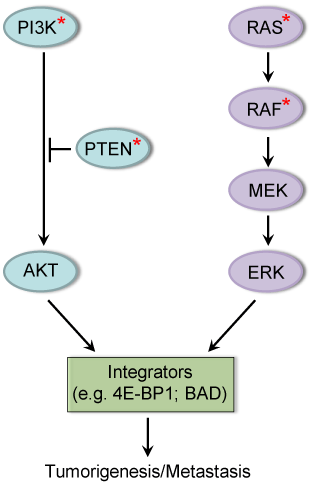
Figure 1 A cross-talk between the PI3K/AKT and RAS/RAF/MEK/ERK signaling pathways via convergent regulation of their common downstream targets for cancer progression. We have identified 4E-BP1 and BAD as key effectors of the oncogenic activation of both pathways that integrate their function in tumorigenesis and metastasis. *mutation
×
AKT and ERK pathways converge on cap-dependent translation
The cap-dependent translation is a process by which most capped mRNAs are translated into proteins. Translation of certain key oncogenic mRNAs bearing long and highly structured 5'-untranslated regions critically depends on the mRNA 5'-cap binding protein eIF-4E. eIF4E is a rate-limiting component of the translation initiation complex eIF4F that also include the scaffolding protein eIF4G and the RNA helicase eIF4A [19]. Consequently, these oncogenic mRNAs are preferentially and disproportionately affected by eIF4E availability and are sensitive to the alteration in the levels of eIF4E [20-22]. The levels of free eIF4E can be increased substantially in cancer cells by a number of mechanisms, including increased eIF4E expression and release of eIF4E from its binding proteins, 4E-BPs, by inactivating phosphorylation of 4E-BPs. 4E-BP1 is a member of the 4E-BP family that represses translation by competing with eIF4G for binding to eIF4E, thereby preventing formation of the eIF4F complex, which recruits the mRNA to the ribosome.
4E-BP1 is frequently hyperphosphorylated and thus inactivated in cancer. The mTOR kinase complex 1 (mTORC1) is a major regulator responsible for 4E-BP1 phosphorylation and activates cap-dependent translation [23]. Both AKT and ERK kinases have been shown to regulate mTORC1 activity via phosphorylation of TSC2 [24,25]. We recently found [15,18] that in colon cancer cells with coexistent mutational activation of AKT and ERK pathways, 4E-BP1 phosphorylation is not affected by inhibition of either pathway alone. However, combined inhibition of both pathways effectively inhibits 4E-BP1 phosphorylation, which in turn activates 4E-BP1 binding to the eIF4E-mRNA cap complex and thus represses eIF4E-initiated cap-dependent translation attendant with the profound suppression of colon tumor growth and metastasis. In addition, a non-phoshorylated and constitutively active mutant 4E-BP1 allele that blocks eIF4E activity exerts similar inhibitory effects on tumor growth and metastasis as the combined inhibition of AKT and ERK pathways in colon cancer, whereas 4E-BP1 knockdown or eIF4E overexpression reduces dependence of colon tumors on AKT and ERK signaling. Mechanistically, we identify that survivin is a key translationally-regulated target of both AKT and ERK pathways through convergence on the mTORC1/4E-BP1/eIF4E axis, and continuous translation of survivin by AKT and ERK signaling is crucial for colon cancer progression to metastasis [18]. These findings highlight 4E-BP1 or eIF4E-initiated cap-dependent translation as a key effector or downstream process of the oncogenice activation of the AKT and ERK pathways triggered tumorigenesis and metastasis.
AKT and ERK pathways converge on survival signals
AKT and ERK signaling pathways have been shown to co-regulate several proteins that promote cancer cell survival and tumor growth. We have previously identified that the BAD protein acts as a switch that integrates the antiapoptotic effects of the PI3K/AKT and MEK/ERK signaling pathways in tumors such as triple-negative breast cancer and glioblastoma in which PI3K/AKT signaling is activated by PTEN loss and MEK/ERK signaling is activated by overexpressed EGFR [26]. BAD is a BH3-domain protein that induces apoptosis by dimerizing with and inactivating the pro-survival proteins Bcl-2 and Bcl-XL[27]. However, in tumors as indicated above, the activated ERK signaling phosphorylates BAD on Ser112, and AKT phosphorylates BAD on Ser136. Phosphorylation of either site is sufficient to be recognized by 14-3-3 proteins, and the 14-3-3 binding prevent apoptosis by sequestering BAD in the cytosol away from the mitochondria and pro-survival Bcl-2 family members. In these cancer cells, inhibition of MEK/ERK pathway has minor effect on cell survival because of phosphorylation of BAD on Ser136 by the constitutively activated PI3K/AKT pathway. Induction of PTEN expression inhibits the PI3K/AKT pathway, but is ineffective to induce apoptosis because of the continued activated MEK/ERK signaling that results in BAD phosphorylation at Ser112. Combined inhibition of the PI3K/AKT and MEK/ERK pathways inhibits BAD phosphorylation on both Ser112 and Ser136, thus releasing BAD from 14-3-3 and inducing massive, BAD-dependent apoptosis associated with synergistic suppression of tumor growth. These data further demonstrate the functional importance of pathway interactions that allows the development of rational strategies for combination therapy.
Clinical perspectives
Several small-molecule inhibitors targeting components of the PI3K/AKT and RAS/RAF/MEK/ERK signaling pathways have been tested in a number of preclinical and clinical studies for the treatment of cancer, but have shown only limited activity as a single agent [28-32]. Coexisting mutational activation of the two pathways could facilitate the development of resistance to therapeutics targeting only one pathway due to pathway convergent regulation on the function of same downstream targets. Combined inhibition of both pathways has been successful in repressing tumor growth in xenograt cancer models and genetically engineered mouse models [15,17,33,34], which may be associated with requirement of inhibition or activation of function of their common targets. Thus, genotyping patient's signaling signatures based on the known activation of PI3K/AKT and MEK/ERK pathways will be important to predict the anti-tumor activity of the pathway inhibitors and to optimize clinical care in the future development of personalized therapy.
Our studies also indicate that direct inhibition of a key output of AKT and ERK signaling pathways may provide an alternative and viable therapeutic strategy to targeting both signaling molecules. For example, given the importance of 4E-BP1-regulated cap-dependent translation in integrating the effects of AKT and ERK on tumor growth and metastasis, compounds that directly mimic 4E-BPs biochemical function or target other translation initiation components have recently produced encouraging anti-tumor effects with limited toxicity profiles in mouse [35-38]. mTOR kinase inhibitors that effectively inhibit the phosphorylation of 4E-BPs may serve as an alternative to the combination of AKT and ERK inhibitors. However, mTOR inhibitors including the active site inhibitors release the feedback inhibition of receptor tyrosine kinases and activate both PI3K/AKT and ERK signaling in tumors [39-41]. Combined inhibition of both AKT and ERK prevents reactivation of AKT and ERK and thus may have different biologic effects and a therapeutic advantage. Identification of more key effectors (integrators) of AKT and ERK signaling pathways could lead to better understanding cancer biology of the pathway interactions and to development of novel biomarkers and drug targets with the potential to change existing treatment paradigms for cancer patients.
Acknowledgments
This work was supported by the Markey Cancer Center Start-up fund and grants from ACS IRG85-001-22, NIH/NCATS UL1RR033173-KL2RR0033171 and NCI R01CA175105 to Q-B She.
References
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009; 8: 627-644.
- Vakiani E, Solit DB. KRAS and BRAF: drug targets and predictive biomarkers. J Pathol. 2011; 223: 219-229.
- Simi L, Pratesi N, Vignoli M, Sestini R, Cianchi F, Valanzano R, et al. High-resolution melting analysis for rapid detection of KRAS, BRAF, and PIK3CA gene mutations in colorectal cancer. Am J Clin Pathol. 2008; 130: 247-253.
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487: 330-337.
- Hou P, Liu D, Shan Y, Hu S, Studeman K, Condouris S, et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin Cancer Res. 2007; 13: 1161-70.
- Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004; 122: 337-341.
- Janku F, Lee JJ, Tsimberidou AM, Hong DS, Naing A, Falchook GS, et al. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS One. 2011; 6: e22769.
- She QB, Chandarlapaty S, Ye Q, Lobo J, Haskell KM, Leander KR, et al. Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS One. 2008; 3: e3065.
- Tanaka H, Yoshida M, Tanimura H, Fujii T, Sakata K, Tachibana Y, et al. The selective class I PI3K inhibitor CH5132799 targets human cancers harboring oncogenic PIK3CA mutations. Clin Cancer Res. 2011; 17: 3272-3281.
- Janku F, Wheler JJ, Naing A, Falchook GS, Hong DS, Stepanek VM, et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013; 73: 276-284.
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364: 2507-2516.
- Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012; 367: 107-114.
- Poulikakos PI, Rosen N. Mutant BRAF melanomas--dependence and resistance. Cancer Cell. 2011; 19: 11-15.
- Yu K, Toral-Barza L, Shi C, Zhang WG, Zask A. Response and determinants of cancer cell susceptibility to PI3K inhibitors: combined targeting of PI3K and Mek1 as an effective anticancer strategy. Cancer Biol Ther. 2008; 7: 307-315.
- She QB, Halilovic E, Ye Q, Zhen W, Shirasawa S, Sasazuki T, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010; 18: 39-51.
- Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009; 69: 4286-4293.
- Halilovic E, She QB, Ye Q, Pagliarini R, Sellers WR, Solit DB, et al. PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res. 2010; 70: 6804-6814.
- Ye Q, Cai W, Zheng Y, Evers BM, She QB. ERK and AKT signaling cooperate to translationally regulate survivin expression for metastatic progression of colorectal cancer. Oncogene. 2013; .
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009; 136: 731-745.
- Mamane Y, Petroulakis E, Martineau Y, Sato TA, Larsson O, Rajasekhar VK, et al. Epigenetic activation of a subset of mRNAs by eIF4E explains its effects on cell proliferation. PLoS One. 2007; 2: e242.
- Livingstone M, Atas E, Meller A, Sonenberg N. Mechanisms governing the control of mRNA translation. Phys Biol. 2010; 7: 021001.
- Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010; 10: 254-266.
- Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006; 25: 6416-6422.
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002; 10: 151-162.
- Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005; 121: 179-193.
- She QB, Solit DB, Ye Q, O'Reilly KE, Lobo J, Rosen N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell. 2005; 8: 287-297.
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995; 80: 285-291.
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009; 9: 550-562.
- Garrido-Laguna I, Hong DS, Janku F, Nguyen LM, Falchook GS, Fu S, et al. KRASness and PIK3CAness in patients with advanced colorectal cancer: outcome after treatment with early-phase trials with targeted pathway inhibitors. PLoS One. 2012; 7: e38033.
- Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013; 10: 143-153.
- Little AS, Smith PD, Cook SJ. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene. 2013; 32: 1207-1215.
- Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol. 2013; 71: 1395-1409.
- Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008; 14: 1351-1356.
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009; 41: 544-552.
- Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007; 128: 257-267.
- Cencic R, Carrier M, Galicia-Vázquez G, Bordeleau ME, Sukarieh R, Bourdeau A, et al. Antitumor activity and mechanism of action of the cyclopenta[b]benzofuran, silvestrol. PLoS One. 2009; 4: e5223.
- Cencic R, Hall DR, Robert F, Du Y, Min J, Li L, et al. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc Natl Acad Sci U S A. 2011; 108: 1046-1051.
- Martineau Y, Azar R, Bousquet C, Pyronnet S. Anti-oncogenic potential of the eIF4E-binding proteins. Oncogene. 2013; 32: 671-677.
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006; 66: 1500-1508.
- Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E, et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011; 1: 248-259.
- Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008; 118: 3065-3074.
Cite this article: Ye Q, She QB (2013) Integration of AKT and ERK Signaling Pathways in Cancer: Biological and Therapeutic Implications. J Pharmacol Clin Toxicol 1(2): 1009.
Current Issue Vol.1.1
The Redox Brain and Nitric Oxide: Implications for Psychiatric Illness
Caroline E Wass1 and Ana Andreazza2,3*
Caroline E Wass1 and Ana Andreazza2,3*
Pharmacological Modulators of Endothelial Progenitor Cell Therapy: Implications for Treatment with Thiazolidinediones
Maria Korah1, Yagna PR Jarajapu2, Maria B. Grant3 and Ashay D. Bhatwadekar3*
Maria Korah1, Yagna PR Jarajapu2, Maria B. Grant3 and Ashay D. Bhatwadekar3*
Profile of Suspect Adverse Drug Reactions in a Teaching Tertiary Care Hospital
Shubhatara Swamy1, Bhanuprakash2, Pratibha Nadig1*, Muralimohan3 and Manjula Shetty1
Shubhatara Swamy1, Bhanuprakash2, Pratibha Nadig1*, Muralimohan3 and Manjula Shetty1
Enhancing the Potency of F508del Correction: A Multi-Layer Combinational Approach to Drug Discovery for Cystic Fibrosis
Emily F Kirby, Ashley S Heard and X Robert Wang*
Emily F Kirby, Ashley S Heard and X Robert Wang*
Leptin Up-Regulates HECTD1 to Promote Phosphoinositide Metabolism and Cell Migration and Invasion in Breast Cancer Cells
Qiaodan Zheng1,2#, Xiang Li2#, Manjula Sunkara3, Andrew J. Morris3, Wenyuan Wu1* and Cai Huang2,4*
Qiaodan Zheng1,2#, Xiang Li2#, Manjula Sunkara3, Andrew J. Morris3, Wenyuan Wu1* and Cai Huang2,4*
Copyright © 2013 JSciMed Central. All rights reserved.